Enfermedad pulmonar intersticial. Revisión
La enfermedad pulmonar intersticial típicamente se presenta con disnea de esfuerzo y puede progresar a insuficiencia respiratoria. El tratamiento de primera línea incluye nintedanib o pirfenidona para la fibrosis pulmonar idiopática y micofenolato de mofetilo para la enfermedad pulmonar intersticial (EPI) debida a una enfermedad del tejido conectivo. Se debe considerar el trasplante de pulmón en pacientes con EPI avanzada. En pacientes con EPI, el entrenamiento físico mejora la distancia de la prueba de caminata de 6 minutos y la calidad de vida. JAMA. 22 de abril de 2024
Importancia La enfermedad pulmonar intersticial (EPI) consiste en un grupo de trastornos pulmonares caracterizados por inflamación y/o fibrosis del parénquima pulmonar asociada con disnea progresiva que frecuentemente resulta en insuficiencia respiratoria terminal. En Estados Unidos, la EPI afecta aproximadamente a 650.000 personas y causa aproximadamente entre 25.000 y 30.000 muertes al año.
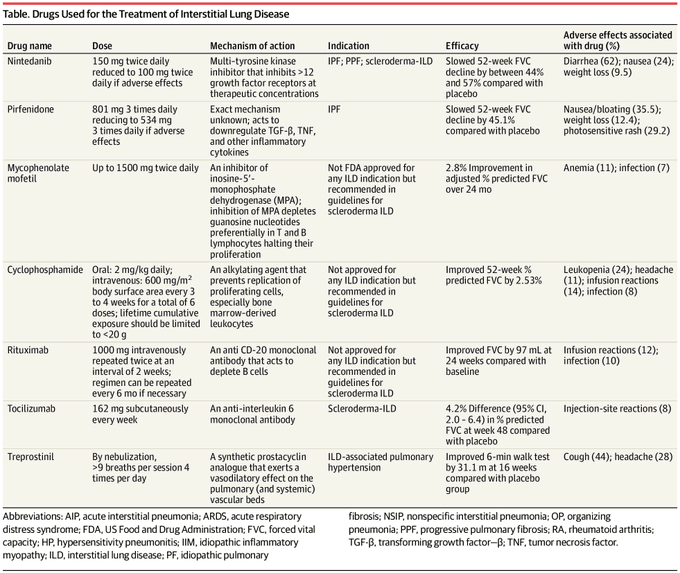
Observaciones Las formas más comunes de EPI son la fibrosis pulmonar idiopática (FPI), que representa aproximadamente un tercio de todos los casos de EPI, la neumonitis por hipersensibilidad, que representa el 15% de los casos de EPI, y la enfermedad del tejido conectivo (ETC), que representa el 25% de los casos. % de casos de EPI. La EPI suele presentarse con disnea de esfuerzo. Aproximadamente el 30% de los pacientes con EPI informan tos. La tomografía computarizada torácica tiene aproximadamente un 91% de sensibilidad y un 71% de especificidad para diagnosticar subtipos de EPI como la FPI. La evaluación fisiológica proporciona información pronóstica importante. Una disminución del 5% en la capacidad vital forzada (FVC) durante 12 meses se asocia con un aumento de aproximadamente el doble de la mortalidad en comparación con ningún cambio en la FVC. La terapia antifibrótica con nintedanib o pirfenidona ralentiza la disminución anual de la FVC en aproximadamente un 44 % a un 57 % en personas con FPI, EPI asociada a esclerodermia y en aquellos con fibrosis pulmonar progresiva de cualquier causa. Para la EPI asociada a enfermedad del tejido conjuntivo, el tratamiento inmunomodulador, como tocilizumab, rituximab y micofenolato de mofetilo, puede retardar la disminución o incluso mejorar la CVF a los 12 meses de seguimiento. La terapia de ejercicio estructurado reduce los síntomas y mejora la distancia de la prueba de caminata de 6 minutos en personas con disnea. El oxígeno reduce los síntomas y mejora la calidad de vida en personas con EPI que tienen una desaturación inferior al 88 % en una prueba de caminata de 6 minutos. El trasplante de pulmón puede mejorar los síntomas y resolver la insuficiencia respiratoria en pacientes con EPI en etapa terminal. Después del trasplante de pulmón, los pacientes con EPI tienen una mediana de supervivencia de 5,2 a 6,7 años en comparación con una mediana de supervivencia de menos de 2 años en pacientes con EPI avanzada que no se someten a un trasplante de pulmón. Hasta el 85% de las personas con EPI fibrótica terminal desarrollan hipertensión pulmonar. En estos pacientes, el tratamiento con treprostinil inhalado mejora la distancia recorrida y los síntomas respiratorios.
Conclusiones y relevancia La enfermedad pulmonar intersticial típicamente se presenta con disnea de esfuerzo y puede progresar a insuficiencia respiratoria. El tratamiento de primera línea incluye nintedanib o pirfenidona para la fibrosis pulmonar idiopática y micofenolato de mofetilo para la enfermedad pulmonar intersticial (EPI) debida a una enfermedad del tejido conectivo. Se debe considerar el trasplante de pulmón en pacientes con EPI avanzada. En pacientes con EPI, el entrenamiento físico mejora la distancia de la prueba de caminata de 6 minutos y la calidad de vida.
La revisión
Maher TM. Interstitial Lung Disease: A Review. JAMA. Published online April 22, 2024. doi:10.1001/jama.2024.3669
Disponible en : https://n9.cl/a5g9y